1.Introduction
The following is intended as documentation for the RaptorX structure alignment server. For details on our structure prediction server, RaptorX, consult http://raptorx.uchicago.edu/documentation/
2.Job Submission
Job submission is done by clicking "New Job" in the top menu of this page. This will display a form (depicted below) through which the user can submit protein structures for pairwise and/or multiple alignment.
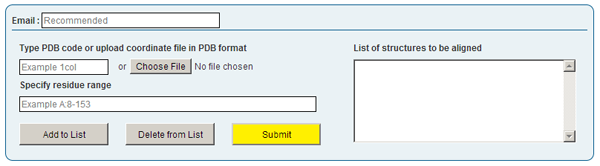
Currently, at most 25 structures are allowed. Structures to be aligned can be added to the list by typing a PDB ID or by uploading a PDB file, followed by clicking "Add to list".
After the "Submit" button is clicked, a unique URL containing the JobID is provided to the user, which can be used to retrieve the structure alignment result later.
You are not required to provide an email address. However, if you do so, the server will email the job result page and a zipped result package after the job is done. This will faciliate job retrieval when the JobID is not kept or lost.
3.Result Interpretation
RaptorX structure alignment server uses a few measures including Lali, RMSD, uGDT(GDT), and TMscore to evaluate the quality of a structure alignment.
Lali: length of alignment. For multiple structure lignment, Lali is the length of core, which consists of all the fully-aligned columns.
RMSD: root-mean-square deviation. For multiple alignment, RMSD is calculated only on the core residues.
uGDT(GDT): uGDT is the unnormalized GDT (Global Distance Test) score defined as 1*N(1)+0.75*N(2)+0.5*N(4)+0.25*N(8), where N(x) is the number of aligned positions with RMSD (in angstrom) smaller than x. GDT is calculated as uGDT divided by the shorter (shortest) protein length and multiplied by a 100.
TMscore: See http://bioinformatics.oxfordjournals.org/content/26/7/889.abstract for an explanation. For multiple structure alignment (MSA), TMScore is the average TMscore of all the pairwise alignments in the MSA. TMscore is between 0 to 1. If TMscore >0.6, it is very likely (90% of chance) that two proteins share a similar fold. When TMscore <0.4, it is very likely (90% of chance) that two proteins have different folds.